Drug Development
Updated on 2023-08-29T11:59:25.152788Z
What is Drug Development?
Drug development is the procedure of getting a novel treatment, therapy, or a drug, to the market after identification of a lead compound.
Pharmaceutical regulatory authorities play a significant role throughout the process of drug development. These authorities are designed to make sure the safety, efficacy, accessibility & security of approved drugs. Taking an example of the United States, the United States Food and Drug Administration (FDA) is a regulatory body that is responsible for the approval of the drug candidate post the drug discovery and development process.
What are the Various Stages of Drug Development?
Process of drug development comprises five key stages- Drug Discovery & Development; Preclinical research; Investigational New Drug Application; Clinical Research; and Regulatory Review, Approval & Post-marketing Surveillance.
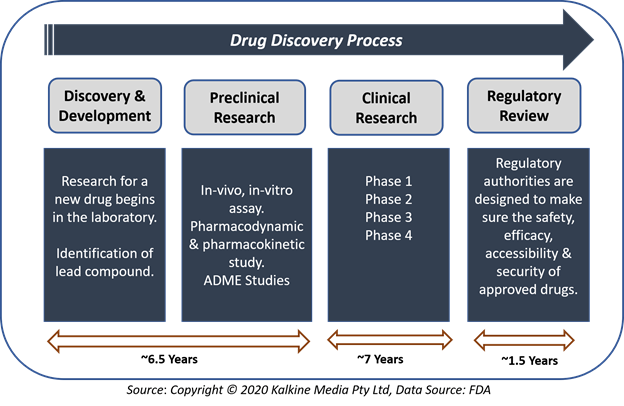
- Drug Discovery & Development
Drug discovery is the process of finding new, innovative medication. During the process of drug development, there may be thousands of compounds as potential candidates for development as a treatment or therapy. After the initial assessment, though, only a few compounds show promising activity and call for further study.
After identification of a promising drug compound for development, the researchers conduct experiments to collect information related to the absorption, distribution, metabolism, and excretion (ADME). Researchers will also find the potential benefit, risk, side effects and adverse events along with the mechanism of action of that specific compound.
- Preclinical Research
Before assessing a new drug in human subjects, scientists must find out whether it has the potential to cause severe harm, also known as toxicity. Preclinical research is of two types: in-vitro and in-vivo.
The research is designed to provide essential information related to the safety and efficacy of a novel drug candidate before its testing in human volunteers. Both in-vitro and in-vivo models are typically used to provide evidence of the biological effect of a new drug candidate. Preclinical trials are required by regulatory authorities such as the FDA and MHRA before submitting an IND (investigational new drug application), which is needed to move to the clinical research.
In preclinical studies, pharmacodynamics and pharmacokinetics of a novel drug candidate are addressed. It is crucial that the most suitable animal models are used to evaluate initial safety information of drug during the preclinical study.
FDA requires scientists to make use of GLP (good laboratory practices) for preclinical laboratory studies.
Generally, preclinical research is not very extensive. However, preclinical studies must give detailed information on drug dosing as well as toxicity levels. After completion of preclinical testing, scientists review their outcomes and determine whether the drug should be tested in humans.
Investigational New Drug Application or IND
The FDA divides investigational new drugs (INDs) into three different types-
- An Investigator IND is submitted by a doctor who initiates as well as investigates. Under the doctor’s direction, the investigational drug is administered. An investigator or physician might submit a research IND to study an unapproved or a new drug.
- In Emergency Use IND the FDA authorises the use of a new investigational drug in an emergency that does not allow time for submission of an IND in accordance with 21CFR (Sec. 312.23 or Sec. 312.20). It is also applied for patients who do not fulfil the standards of the current study protocol, or if there is no existing approved study protocol.
- Treatment IND is for experimental drugs which show promise in clinical testing for severe or immediately life-threatening diseases while the final clinical work is conducted, and the FDA review takes place.
After submission of the IND, the investigator or sponsor must wait for thirty days before starting any clinical research. During this one month, the FDA review the IND for safety to make sure that research subjects will not be subjected to any unreasonable risk.
The IND application should comprise information in three areas, including animal pharmacology & toxicology studies, manufacturing details, along with the information related to clinical protocols & study investigator.
Drug developing companies are free to seek help from the FDA at any point in the process of drug development, including:
- Pre-IND application for the review of FDA guidance documents as well as find answers for augmenting their research.
- After Phase 2 clinical trial, to obtain guidance on the design of large Phase 3 clinical trials.
- During the process to get an evaluation of the IND application.
IND Approval
The FDA review team has thirty days for the review of the original IND submitted by the drug developer. This process protects the clinical trial participants from any substantial and unnecessary risk during clinical trials. For the IND applications, the US regulator provides its response in two aspects:
- Approval to commence the clinical trials.
- Clinical hold for halt or delay of the examination.
For the clinical hold, the FDA can put the trial on hold for specific causes, including:
- Trial subjects are exposed to significant or any unreasonable risk.
- Investigators of the clinical are not qualified.
- Materials for the study volunteers are misleading.
- If the IND application does not comprise sufficient information related to the risks of the clinical trial.
However, a clinical hold is unusual, and as an alternative to the hold, FDA often offers comments to improve the clinical trial quality.
In most instances, if the regulatory agency is convinced that the clinical trial fulfils the Federal standards, the applicant is permitted to continue the proposed clinical study.
Moreover, the drug developer is accountable for informing the FDA review team about new protocols, along with the serious side effects observed during the study. This information makes sure that the review team can monitor the clinical trials thoroughly to signal any side effects. After the completion of the clinical trial, investigators must submit the reports of the study.
The process of informing the FDA about the trial’s new protocols continues until the applicant decides to end clinical trials or files a marketing application. Before filing a marketing application, the drug developer must have sufficient information from two large, controlled clinical trials.
- Clinical Research
Several types of clinical research are done depending on what research is going on. Some types of clinical research are-

Source: Copyright © 2020 Kalkine Media Pty Ltd, Data Source: FDA
Other types of clinical research
Other than the above-mentioned research, there are some other clinical studies that do not involve drug investigation, and a person’s regular medications may not require to be altered.
Besides, healthy volunteers are also needed to compare their results to the results of patients being studied.
Below are a few examples of other kinds of research:
- Long-term clinical research that includes brain scans or psychological tests.
- A genetic study, including blood tests but without any change in medication.
- A clinical study of family history involving family members to learn about medical needs and history of people.
When clinical research is used for evaluation of any medication or device, it is conducted in four clinical trial phases-
- Phase 1 Clinical trials- In Phase 1, researchers test an investigational drug or treatment in a small group of volunteers for the first time. Safety and efficacy of the drug are evaluated in this phase with the identification of side effects.
- Phase 2 Clinical trials- In this phase, an experimental drug is given to a larger group of subjects to see if it is effective and to evaluate its safety further.
- Phase 3 Clinical trials- The experimental study drug or treatment is given to large groups of people. Researchers confirm the effectiveness of the new drug, monitor side effects, compare it to commonly used medications, and collect information that will permit the experimental drug or treatment to be used safely.
- Phase 4 Clinical trials- Phase 4 clinical trial comprises post-marketing studies that are organized after approval of a drug or therapy for use by the FDA. Post-marketing studies provide some additional information, including the risks, adverse effect and best use associated with the treatment.
To Know More About Clinical Trials Click Here.
New Drug Application or NDA
An NDA reveals the complete story of a drug, and its objective is to determine the safety and efficacy of the drug for its anticipated application in the population studied.
In an NDA, a drug developer must incorporate every information related to a drug, from preclinical study data to Phase 3 clinical trial data. With the results of the clinical trials, drug developers should include the below-mentioned information-

On receiving an NDA, the FDA review team determines whether it is complete or not. If the application is incomplete, the review team has the right to reject to file the New Drug Application. If it is complete, the team has six to ten months to decide the drug approval.
In instances where the FDA finds that a drug has been demonstrated safe and effective for its intended use, then the agency will work with the applicant for the development as well as refining of the prescribing information. This process is termed as labeling. Labeling precisely and accurately describes the basis for approval and the best use of the drug.
- Regulatory Review, Approval & Post-marketing Surveillance
Suppose a drug manufacturing company has evidence from its preliminary tests, preclinical and clinical research that the innovative drug/treatment is safe and effective for its intended use. In that case, the drug developer may file an application for the marketing of the drug.
FDA approval for a medicine/drug means that the Center for Drug Evaluation and Research (CDER) has reviewed the information on the effects shown by the new drug, and during the review, the drug provided benefits outweighing its known risks for the intended population.
If clinical trials provide vital information on the safety and efficacy of a drug, it is not possible to have complete information related to the safety of a drug at the time of approval. Hence, the FDA reviews reports of problems with prescription and over-the-counter (OTC) drugs. Based on these reports, the FDA decides to add cautions to the dosage or the usage information of the drug, along with some other measures related to serious issues or adverse effects.
Post-marketing safety surveillance is the monitoring of a drug after receiving the approval and market launch. The surveillance is designed for the evaluation of long-term safety along with the drug efficacy.
Besides, the FDA is developing a new national system to spot potential safety issues more swiftly under the Sentinel Initiative. This system will utilize extremely large existing electronic health databases such as electronic health records (EHR) systems, administrative and insurance claims databases, and registries to track approved drugs’ safety in real-time.
According to the FDA. This tool will be an addition to the existing post-market safety assessment tools of the FDA.
Drug development is a time, and knowledge-intensive process and several healthcare companies outsource the entire process, or parts of it, to organizations known as Contract Manufacturing Organizations (or CMOs). Click here to learn about one such Australian player, Sypharma.